
Abstrak
Pembelajaran mesin (ML) telah digunakan untuk memprediksi energi aktivasi untuk reaksi-reaksi utama dalam proses pergeseran gas air pada permukaan MXene murni, yaitu, disosiasi air (H 2 O) dan hidroksil (OH), dan reaksi pembentukan hidrogen (H 2 ) dan karbon dioksida (CO 2 ). Set data mencakup 92 MXene, termasuk komposisi logam transisi tunggal (M 2 N atau M 2 C) dan logam transisi ganda (M’ 2 M”C 2 ), dengan energi yang berasal dari kalkulasi teori fungsi kerapatan dari karya ini atau literatur. Beberapa model ML dievaluasi, termasuk Random Forest Regressor (RFR), Gradient Boosting Regressor, Jaringan Syaraf Tiruan, Support Vector Machines, Decision Tree Regressor, dan K-Nearest Neighbors Regressor, yang mengidentifikasi RFR sebagai yang paling akurat. Plot korelasi yang membandingkan energi aktivasi aktual versus yang diprediksi selanjutnya memvalidasi kekokohan dan keakuratan model kami. Analisis kepentingan fitur mengungkap bahwa dua faktor kunci, energi reaksi dan koefisien partisi logaritmik (LogP) reaktan, sangat penting dalam memprediksi energi aktivasi. Sementara fitur pertama selaras dengan prinsip Bronsted-Evans-Polanyi yang berkaitan dengan kinetika dan termodinamika reaksi tertentu, fitur kedua mencakup berbagai sifat fisikokimia hanya dalam satu parameter: polaritas, ikatan hidrogen, ukuran molekul, bentuk molekul, dan muatan. Studi ini menggarisbawahi kekuatan ML dalam katalisis, menawarkan kerangka kerja prediktif untuk memandu desain katalis.
1 Pendahuluan
Kebutuhan akan solusi energi yang berkelanjutan dan efisien telah mendorong minat pada bahan yang mampu mengkatalisis reaksi kimia utama. MXenes, kelas karbida dan nitrida logam transisi dua dimensi (2D), telah muncul sebagai kandidat yang menjanjikan karena sifat fisikokimianya yang khas [ 1 – 5 – 6 ]. Sejak ditemukan pada tahun 2011, mereka telah menarik perhatian karena konduktivitas listriknya yang tinggi, hidrofilisitas, dan kimia permukaan yang dapat disetel, yang memungkinkan aplikasi dalam penyimpanan energi, penginderaan, dan katalisis [ 7 – 10 – 11 ]. MXenes disintesis dengan mengetsa secara selektif lapisan-A dari fase MAX, yang merupakan karbida terner berlapis atau nitrida dengan rumus umum M n + 1 AX n , di mana n adalah bilangan bulat (1–4), M adalah logam transisi awal, A biasanya merupakan unsur dari golongan 13 atau 14 dari Tabel Periodik, dan X adalah karbon dan/atau nitrogen) [ 12 – 14 – 15 ]. Struktur 2D yang dihasilkan biasanya mempertahankan sifat metalik dari logam transisi sambil menunjukkan reaktivitas permukaan yang ditingkatkan karena gugus terminal seperti -OH, -O, dan -F [ 16 , 17 – 18 ]. Struktur unik ini menyediakan area permukaan yang luas dan situs aktif yang melimpah, yang penting untuk reaksi katalitik [ 19 , 20 ]. Selain itu, kimia permukaan MXene yang dapat disetel memungkinkan modifikasi sifat elektroniknya, mengoptimalkan aktivitas katalitik untuk reaksi tertentu. Kemampuan penyetelan ini sangat penting dalam reaksi yang melibatkan molekul kecil seperti H 2 , OH, H 2 O, dan CO 2 , yang merupakan pusat teknologi konversi dan penyimpanan energi, karena reaktivitas dan interaksinya yang unik dengan katalis.
Produksi dan penyimpanan hidrogen, pemisahan air, dan reduksi CO 2 adalah proses penting dalam sistem energi terbarukan [ 21 , 22 – 23 ]. Hidrogen, sebagai pembawa energi bersih, memerlukan katalis yang efisien untuk produksinya (melalui pemisahan air) dan penyimpanannya [ 24 , 25 ]. Demikian pula, mengubah CO 2 menjadi bahan kimia dan bahan bakar yang berguna mengatasi masalah pasokan energi dan lingkungan. MXenes, dengan konduktivitas tinggi dan kepadatan situs permukaan aktif, menawarkan platform yang menjanjikan untuk proses katalitik ini [ 26 – 29 – 30 ]. Memahami interaksi antara MXenes dan adsorbat pada tingkat atom sangat penting untuk mengoptimalkan kinerja katalitik. Energi aktivasi, yang menentukan laju reaksi kimia, merupakan parameter penting dalam konteks ini. Kemampuan untuk memprediksi energi aktivasi secara akurat dapat secara signifikan meningkatkan desain dan pengembangan katalis berbasis MXene [ 31 ].
Kemajuan terbaru dalam pembelajaran mesin (ML) telah merevolusi berbagai bidang, termasuk ilmu material [ 32 , 33 – 34 ]. Teknik ML dapat mengungkap pola dan hubungan yang kompleks dalam kumpulan data yang besar, memungkinkan prediksi yang sangat akurat tentang sifat dan kinerja material [ 35 , 36 ]. Dalam katalisis, model ML dapat memprediksi energi aktivasi, laju reaksi, dan mengidentifikasi deskriptor utama yang memengaruhi aktivitas katalitik [ 37 – 39 – 40 ]. Misalnya, Roy et al. [ 41 ] menggunakan pembelajaran mesin untuk memprediksi fungsi kerja MXenes. Mereka mengembangkan model menggunakan sifat kimia dasar sebagai fitur, dilatih pada 275 titik data dari Computational 2D Materials Database. Model jaringan saraf mereka mencapai kesalahan absolut rata-rata (MAE) sebesar 0,12 eV pada data pelatihan dan 0,25 eV pada data pengujian. Analisis kepentingan fitur mengungkapkan bahwa elektronegativitas atom-atom yang berakhir di permukaan sangat memengaruhi fungsi kerja. Model orde tereduksi dengan fitur yang lebih sedikit menunjukkan transferabilitas yang efektif ke material baru, memprediksi fungsi kerja MXenes dengan terminasi permukaan baru seperti Br, Cl, S, N, dan NH. Pendekatan ini memfasilitasi identifikasi cepat MXenes yang disesuaikan dengan sifat elektronik yang ditargetkan untuk aplikasi tertentu. Demikian pula, Abraham et al. [ 42 ] menggabungkan penyaringan mekanika kuantum throughput tinggi dengan kecerdasan buatan untuk mengidentifikasi deskriptor utama untuk aktivasi CO 2 pada MXenes. Model pembelajaran mesin mereka menyaring 114 MXenes murni dan cacat, dengan model RFR menunjukkan kinerja terbaik dalam memprediksi energi adsorpsi CO 2 . Model RFR menunjukkan kesalahan absolut rata-rata sebesar 0,16 eV untuk data pelatihan dan 0,42 eV untuk data pengujian. Analisis kepentingan fitur mengidentifikasi pusat pita-d, elektronegativitas logam permukaan, dan jumlah elektron valensi atom logam sebagai deskriptor penting. Pendekatan ini menyoroti potensi pembelajaran mesin untuk memandu desain katalis berbasis MXene baru dengan memprediksi indikator utama untuk aktivasi CO2 .
Beberapa penelitian telah mengeksplorasi adsorpsi dan disosiasi molekul seperti H 2 , H 2 O, dan CO 2 pada permukaan MXene. Misalnya, Wang et al. [ 43 ] menyelidiki adsorpsi dan disosiasi hidrogen pada lapisan tunggal Ti 2 C dua dimensi . Meskipun mereka menemukan bahwa hidrogen terdisosiasi secara spontan pada Ti 2 C, penting untuk dicatat bahwa MXene tanpa fungsionalisasi permukaan sulit untuk distabilkan. Selain itu, Tahini et al. [ 44 ] mempelajari adsorpsi air pada V 2 CO 2 , Ti 2 CO 2 , dan Nb 2 CO 2 dan meskipun energi adsorpsi masing-masing adalah -0,23, -0,15 dan -0,49 eV, penambahan energi titik nol dan kontribusi entropi orde 0,67 eV menghasilkan nilai energi Gibbs positif. Namun, penggabungan atom logam tunggal yang dijangkarkan pada MXenes ini, untuk menghasilkan, misalnya, Fe@V 2 CO 2 , Mn@Ti 2 CO 2 , dan Ir@Nb 2 CO 2 , meningkatkan stabilitas air yang diserap, yang berkontribusi pada disosiasi yang relatif mudah. Jurado et al. [ 45 ] memeriksa permukaan MXenes nitrida tanpa terminasi untuk penangkapan CO 2 menggunakan kalkulasi prinsip pertama. MXenes nitrida berbasis Ti-, Hf-, dan Zr menunjukkan adsorpsi paling eksotermik. Parey et al. [ 46 ] menggunakan teori fungsi kerapatan (DFT) untuk menyelidiki potensi MXenes Zr 2 X (OH) 2 (X = C, N, atau B) untuk konversi CO 2 menjadi asam format melalui mekanisme hidroksilasi permukaan. Hal ini melibatkan transfer proton–elektron berpasangan dua tahap, di mana CO 2 dihidrogenasi menggunakan atom hidrogen dari permukaan MXene berujung OH. Hasil mereka menunjukkan bahwa abstraksi hidrogen dari permukaan berlangsung mudah, secara efektif melewati batasan termodinamika.
Bahasa Indonesia: Untuk mempercepat penemuan di luar metode DFT tradisional, beberapa studi terkini telah menggunakan pembelajaran mesin untuk mengeksplorasi potensi katalitik MXenes. Misalnya, Yang et al. [ 47 ] menggabungkan ML dengan DFT throughput tinggi untuk menyelidiki MXenes berbasis V 2 C yang didoping non-logam (yaitu, P, S, Se, Te) dengan terminasi permukaan yang berbeda (O, S). Temuan mereka menunjukkan bahwa modifikasi yang diinduksi dopan dalam kerapatan elektron orbital p z secara signifikan memengaruhi energi bebas Gibbs dari adsorpsi hidrogen (Δ G H* ), yang memungkinkan prediksi aktivitas reaksi evolusi hidrogen (HER) yang akurat di berbagai komposisi. Mereka mengembangkan deskriptor turunan ML universal ( R 2 > 0,93) yang mengidentifikasi P–V 2 CTe 2 sebagai katalis berkinerja terbaik dengan Δ G H* yang lebih baik daripada Pt dan stabilitas termal yang tinggi [ 47 ]. [ 48 ] memperkenalkan kerangka kerja multilangkah ML/DFT yang kuat untuk menilai aktivitas HER dari lebih dari 4.500 MM′XT 2 -jenis MXenes. Menggunakan regresi peningkatan gradien dengan eliminasi fitur rekursif, model mereka memperkirakan Δ G H* dengan kesalahan absolut rata-rata rendah sebesar 0,358 eV. Khususnya, MXenes berbasis Nb-, Mo-, dan Cr dengan terminasi oksigen muncul sebagai katalis HER yang sangat aktif, melampaui sistem berbasis Pt komersial. Pekerjaan oleh Moses et al. [ 48 ] menggarisbawahi efisiensi ML dalam menavigasi ruang desain komposisi MXenes yang luas untuk pembangkitan hidrogen. Dalam penyaringan ML yang ditingkatkan secara terpisah, Xu et al. [ 49 ] memeriksa 78 MXenes TiVCO multimetal Janus yang didoping 2 , menggunakan model klasifikasi yang dilatih pada nilai Δ G H* yang diturunkan dari DFT . Model ML 7-fitur mereka mencapai akurasi prediksi sebesar 93,6% dan mengidentifikasi lima katalis S-doped berkinerja tinggi, yang kemudian divalidasi oleh DFT. Yang penting, penelitian Xu et al. [ 49 ] ini menggambarkan nilai menggabungkan klasifikasi ML dengan DFT untuk penemuan katalis yang efisien, terutama dalam sistem dengan data pelatihan terbatas.
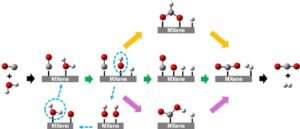
Dalam karya ini, algoritma pembelajaran mesin seperti Random Forest Regressor (RFR), Gradient Boosting Regressor (GBR), Artificial Neural Networks (ANN), Support Vector Machines (SVM), Decision Tree Regressor (DTR), dan K-Nearest Neighbors Regressor (KNR) secara efektif digunakan untuk memodelkan dan memprediksi energi aktivasi reaksi disosiasi dan asosiasi. Model-model ini menangani data berdimensi tinggi, menangkap hubungan nonlinier, dan memberikan wawasan ke dalam mekanisme yang mendasari yang mengatur perilaku material. Tujuan utama dari karya ini adalah untuk menerapkan teknik pembelajaran mesin untuk memprediksi energi aktivasi untuk reaksi disosiasi H 2 O dan OH serta reaksi asosiasi H 2 dan CO 2 pada permukaan MXene. Reaksi-reaksi ini mendasar bagi reaksi pergeseran gas air (WGS) (H 2 O + CO → H 2 + CO 2 , Gambar 1 ), proses penting dalam produksi hidrogen [ 51 – 53 – 54 ]. Memahami energi aktivasi reaksi fundamental ini sangat penting untuk meningkatkan proses katalitik dan merancang katalis yang lebih efisien untuk reaksi WGS.
2 Metodologi
2.1 Kumpulan Data
Dataset yang digunakan dalam studi ini terdiri dari 92 titik data yang mewakili berbagai sifat MXene, termasuk interaksinya dengan molekul yang diinginkan: H 2 O, OH, H 2 , dan CO 2 . Sifat-sifat ini diperoleh dari dua sumber utama: perhitungan DFT untuk 10 MXene (bimetalik), dan data literatur [ 17 , 55 , 56 ] untuk MXene (logam tunggal) yang tersisa. Pada titik ini penting untuk menambahkan bahwa data, baik yang dihitung dalam pekerjaan ini atau diambil dari literatur, dihitung dengan parameter komputasi yang identik (misalnya fungsi korelasi-pertukaran yang sama, batas). Oleh karena itu, potensi kekhawatiran mengenai homogenitas data sebagian besar dikurangi dalam konteks ini.
Dataset lengkap, yang mencakup semua properti relevan, data interaksi, serta kode dan model pembelajaran mesin yang digunakan dalam studi ini, tersedia daring di ( https://doi.org/10.17632/4zmkpw3wxx.2 ). Dataset tersebut mencakup 28 titik data untuk disosiasi H 2 O (H 2 O → HO + H), 18 titik data untuk disosiasi OH (OH → O + H), 28 titik data untuk asosiasi H 2 (2 H → H 2 ), dan 18 titik data untuk pembentukan CO 2 (CO + O → CO 2 ).
Karena aplikasi pembelajaran mesin dalam katalisis berbasis MXene masih dalam tahap awal, studi ini berfokus pada permukaan MXene yang polos (tidak terfungsionalisasi) dan bebas cacat untuk mengisolasi efek intrinsik dari kimia inti logam transisi. Pendekatan ini memungkinkan perbandingan yang sistematis dan tidak bias di berbagai komposisi MXene dengan menghilangkan variabilitas yang disebabkan oleh terminasi dan cacat permukaan. Sementara MXene di dunia nyata sering kali diakhiri dengan gugus fungsional seperti –O, –OH, dan –F, dan dapat menunjukkan ketidaksempurnaan struktural seperti kekosongan atom atau substitusi [ 18 – 59 , 57 – 60 ], mengecualikan faktor-faktor ini memberikan dasar yang berharga untuk mengungkap tren aktivitas fundamental. Kerangka kerja yang terkontrol ini memastikan konsistensi data, yang penting untuk melatih model pembelajaran mesin yang andal dan mengidentifikasi hubungan struktur-properti umum. Di luar kekuatan prediktif hasil kami, pendekatan ini meletakkan dasar bagi penelitian masa depan yang menggabungkan terminasi dan cacat permukaan untuk mengeksplorasi bagaimana fitur ini memodulasi energi aktivasi dan memengaruhi kinerja katalitik dalam kondisi yang relevan secara eksperimen.
2.2 Rincian Komputasi DFT
Untuk 10 MXenes dengan rumus M′ 2 M′′C 2 , di mana M′ adalah Cr, Mo, atau Ti, dan M′′ adalah Nb, Ta, Ti, atau V, perhitungan DFT dilakukan untuk mendapatkan data seperti energi aktivasi, muatan Bader, parameter kisi, dan deskriptor penting lainnya yang terkait dengan MXenes. Parameter ini, yang penting untuk memahami sifat katalitik MXenes, tercantum dalam Tabel S1 dalam Informasi Pendukung. Perhitungan dilakukan dengan menggunakan Vienna Ab Initio Simulation Package (VASP) [ 61 ]. Secara khusus, fungsional korelasi-pertukaran Perdew–Burke–Ernzerhof (PBE) digunakan, sebuah fungsional Generalized Gradient Approximation (GGA) yang digunakan secara luas yang secara efektif mewakili struktur elektronik dan energetika berbagai bahan, termasuk MXenes [ 62 ]. Untuk memperhitungkan interaksi van der Waals jarak jauh, koreksi dispersi D3 disertakan dalam perhitungan [ 63 ]. Pendekatan ini memberikan penanganan fenomena adsorpsi yang kuat yang cocok untuk sistem logam dan non-logam. MXenes yang dipelajari dalam karya ini, dengan rumus M 2 C, M 2 N, dan M’ 2 M”C 2 , dimodelkan dengan 16 atom per lapisan untuk merepresentasikan permukaannya, yaitu, menggunakan struktur supersel periodik p (4 × 4), seperti yang diilustrasikan dalam Gambar 2 , dan grid titik- k 3 × 3 × 1 dalam zona Brillouin [ 64 ]. Molekul yang terisolasi dimodelkan dalam kotak asimetris dengan dimensi 10 × 11 × 12 Å 3 , menggunakan titik- k tunggal ( titik Γ ). Kriteria konvergensi untuk medan konsisten diri (SCF) dan langkah relaksasi ionik ditetapkan pada 10 −6 eV dan 0,005 eV/Å, masing-masing, untuk memastikan akurasi yang memadai. Batas energi gelombang bidang ditetapkan pada 550 eV, nilai standar untuk perhitungan ini. Potensi semu gelombang augmented proyektor (PAW) digunakan untuk semua elemen dalam sistem, dan relaksasi ionik dilakukan menggunakan algoritma gradien konjugat [ 65 ].

di mana E a (disosiasi) adalah energi aktivasi untuk reaksi disosiasi yang sesuai, dan E r (disosiasi) adalah energi reaksi disosiasi. Energi aktivasi yang rendah menunjukkan bahwa permukaan MXene mengkatalisis reaksi, baik pemutusan ikatan (disosiasi) maupun pembentukan ikatan (asosiasi), secara lebih efektif, oleh karena itu permukaan MXene yang sesuai merupakan katalis yang lebih baik.
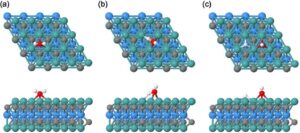
Gambar 3 menyoroti konfigurasi jalur reaksi katalitik yang penting, yaitu, (a) keadaan awal → (b) keadaan transisi → (c) keadaan akhir, menggunakan disosiasi air pada permukaan M’ 2 M”C 2 MXene sebagai contoh. Struktur pada panel (a) menunjukkan struktur molekul air yang dioptimalkan yang berinteraksi dengan permukaan melalui fisisorpsi atau kemisorpsi lemah. Energi adsorpsi air dihitung dari energi total MXene dengan air yang teradsorpsi dan energi fragmen yang terpisah, yaitu molekul H 2 O fase gas dan permukaan model MXene. Panel (b) menggambarkan keadaan transisi, yang mewakili titik energi tertinggi sepanjang koordinat reaksi, tempat ikatan O–H diregangkan. Perbedaan energi antara struktur pada panel (b) dan (a) sesuai dengan energi aktivasi reaksi disosiasi air, dengan permukaan yang lebih aktif memiliki energi aktivasi yang lebih rendah daripada permukaan yang kurang aktif. Panel (c) menyajikan keadaan akhir, dengan fragmen H dan OH yang terdisosiasi teradsorpsi di lokasi yang berbeda. Perbedaan energi antara struktur pada panel (c) dan (a) sesuai dengan energi reaksi (nilai negatif/positif adalah reaksi eksotermik/endotermik).
2.3 Rekayasa Fitur
Bahasa Indonesia: Untuk meningkatkan dataset dan menangkap interaksi kompleks antara MXenes dan adsorbat, kami memperoleh fitur tambahan dengan menggunakan tiga pustaka Python: Matminer, Mendeleev dan RDKit. Matminer digunakan untuk memperoleh fitur material [ 66 ], sedangkan Mendeleev digunakan untuk memperoleh sifat unsur yang terkait dengan atom MXene. Di sisi lain, RDKit [ 67 ], pustaka cheminformatics, digunakan untuk menghitung deskriptor molekuler untuk spesies adsorbat utama dari setiap reaksi, termasuk H 2 O, OH, H 2 , dan CO 2 . Fitur-fitur ini mencakup berbagai kategori, masing-masing diperoleh dengan menggunakan metode yang berbeda. Komposisi kimia dan struktur molekuler material dianalisis untuk memperoleh fitur-fitur seperti afinitas elektron, sifat ionisasi, pusat pita, dan konduktivitas termal menggunakan Matminer. Untuk spesies adsorbat utama dari setiap reaksi (H 2 O, OH, H 2 , dan CO 2 ), sifat-sifat seperti berat molekul, muatan parsial, elektron radikal, dan elektron valensi diperoleh menggunakan RDKit. Karakteristik logam transisi dalam MXenes ditentukan menggunakan Mendeleev, termasuk sifat-sifat unsur seperti jari-jari atom, berat atom, titik leleh, elektronegativitas, dan energi ionisasi pertama. Selain itu, sifat struktural, elektronik, dan termodinamika MXenes diturunkan dari perhitungan DFT, termasuk panjang ikatan, jarak lapisan, muatan Bader, dan energi pembentukan. Sifat permukaan dan reaktivitas juga diselidiki menggunakan DFT, seperti energi adsorpsi reaktan (dalam reaksi yang dijelaskan dalam Persamaan ( 3 ) dan ( 4 ), energi adsorpsi dievaluasi sebagai jumlah CO dan O, atau dua atom H, masing-masing), energi reaksi dan energi aktivasi. Kombinasi metode ini memastikan serangkaian fitur komprehensif yang menangkap sifat-sifat penting yang relevan dengan prediksi energi aktivasi. Secara keseluruhan, kami menghasilkan 103 fitur, yang dirangkum bersama dengan deskripsinya di Tabel S2 dalam Informasi Pendukung, untuk dimasukkan ke dalam model kami. Dengan mengintegrasikan fitur-fitur terhitung ini ke dalam model, model dapat mengenali pola dan hubungan yang kompleks dalam kumpulan data, mengidentifikasi fitur mana yang paling penting dan berpengaruh dalam memprediksi energi aktivasi.
Kami menggunakan eliminasi fitur rekursif (RFE) untuk pemilihan fitur, untuk mengidentifikasi fitur yang paling relevan untuk prediksi, sekaligus menghilangkan fitur yang berlebihan atau kurang informatif. Dengan secara sistematis menghilangkan fitur yang paling tidak penting, RFE menyempurnakan model untuk meningkatkan kinerja dan interpretabilitasnya. Fitur dengan kepentingan yang lebih rendah dibuang, dan model dilatih ulang menggunakan fitur yang tersisa. Proses iteratif ini berlanjut hingga hanya fitur penting yang dipertahankan. Dalam pekerjaan ini, RFE dikombinasikan dengan validasi silang dan kesalahan absolut rata-rata untuk memberi peringkat dan memilih fitur yang paling penting. Kumpulan fitur terakhir, setelah beberapa iterasi, merupakan fitur yang paling penting untuk membuat prediksi yang akurat, sehingga meningkatkan akurasi dan interpretabilitas model kami.
2.4 Model Pembelajaran Mesin
Bahasa Indonesia : Pada Gambar 4 , kami mengilustrasikan alur kerja proses pembelajaran mesin yang digunakan dalam studi ini untuk memprediksi energi aktivasi untuk disosiasi H 2 O, OH, dan untuk reaksi asosiasi H 2 dan CO 2 pada permukaan MXene. Proses dimulai dengan analisis struktur MXene, diikuti oleh pengumpulan data dari kalkulasi DFT dan karya referensi literatur. Data yang terkumpul kemudian diproses melalui rekayasa fitur untuk mengidentifikasi deskriptor utama, dengan korelasi yang divisualisasikan menggunakan peta panas. Model ML selanjutnya dilatih dan dievaluasi untuk memilih model dengan performa terbaik dan hiperparameter yang dioptimalkan menurut validasi silang, yang kemudian diuji untuk akurasi dan generalisasi menggunakan set pengujian akhir dengan data holdout yang digunakan hanya untuk tujuan ini. Alur kerja ini menggarisbawahi integrasi teknik komputasional dan pembelajaran mesin dalam penelitian MXenes, dengan pengembangan model yang dijelaskan secara rinci dalam beberapa subbagian berikutnya.

Dalam studi ini, beberapa model ML digunakan, masing-masing dengan kelebihannya sendiri. RFR digunakan karena kemampuannya untuk menangani dataset besar dan kompleks dan secara efektif menangkap hubungan nonlinier [ 68 ]. ANN dipilih karena kemampuannya untuk memodelkan pola rumit melalui struktur berlapis. ANN sangat cocok untuk data berdimensi tinggi, karena dapat mendeteksi hubungan kompleks antara variabel dengan meniru cara otak manusia memproses informasi [ 69 ]. Untuk lebih meningkatkan kinerja prediksi, GBR dipertimbangkan karena efisiensinya dalam menggunakan teknik boosting. Ia bekerja dengan menggabungkan model yang lebih lemah menjadi model yang lebih kuat dan terkenal karena akurasinya yang tinggi dan kemampuannya untuk menangani data yang bising [ 70 ]. SVM juga diterapkan karena efektivitasnya dalam menemukan hyperplane optimal untuk tugas regresi. SVM dapat menangani data linier dan nonlinier, membuatnya serbaguna untuk berbagai jenis masalah [ 71 ]. DTR disertakan karena sifatnya yang intuitif, memberikan interpretasi yang jelas dengan membagi dataset menjadi cabang-cabang berdasarkan nilai fitur. Meskipun rentan terhadap overfitting, pohon keputusan mudah divisualisasikan dan dipahami [ 72 ]. Selain itu, regresor k-nearest neighbour dipilih karena kesederhanaan dan efektivitasnya, terutama untuk kumpulan data yang lebih kecil. Model ini memprediksi output berdasarkan titik data terdekat, menjadikannya alat yang mudah dipahami tetapi ampuh untuk tugas regresi [ 73 ]. Selain model-model ini, regresi linier multivariabel (MLR) juga diterapkan karena kesederhanaan dan interpretabilitasnya. MLR mengasumsikan hubungan linier antara prediktor dan variabel respons, menjadikannya model dasar yang berharga untuk dibandingkan dengan model pembelajaran mesin yang lebih kompleks [ 74 ].
Dengan mengimplementasikan model-model ini menggunakan pustaka scikit-learn Python [ 75 , 76 ], penelitian ini bertujuan untuk membandingkan dan mengidentifikasi model-model yang paling efektif, yang pada akhirnya memastikan prediksi energi aktivasi yang kuat dan andal dalam konteks reaksi yang dikatalisis MXene. Untuk menjamin evaluasi menyeluruh dan prediksi energi aktivasi yang akurat, kami mengimplementasikan pendekatan terstruktur yang melibatkan pelatihan, validasi, dan pengujian. Model-model tersebut pertama-tama dilatih dan divalidasi melalui validasi silang menggunakan 70% dari kumpulan data (64 titik data), yang memainkan peran penting dalam mengoptimalkan model dan mengurangi overfitting. 30% sisanya (28 titik data) disisihkan sebagai set pengujian untuk mengevaluasi kinerja model pada data yang sebelumnya tidak terlihat.
2.5 Penyetelan dan Evaluasi Model
Dalam pekerjaan ini, kami mengikuti metodologi yang ketat untuk pelatihan dan evaluasi model untuk memastikan keakuratan dan keandalan prediksi energi aktivasi. Setiap model dinilai secara menyeluruh menggunakan validasi silang 5 kali lipat, di mana kumpulan data dibagi menjadi lima subset untuk pelatihan dan validasi berulang. Kami menggunakan beberapa model untuk lebih memahami hubungan antara fitur input dan energi aktivasi. Untuk menyempurnakan model kami, kami menerapkan pengoptimalan hiperparameter menggunakan RandomizedSearchCV, dengan semua detail disediakan dalam Informasi Pendukung. Teknik ini melibatkan pengambilan sampel acak kombinasi hiperparameter dan mengevaluasi kinerjanya. Untuk mengevaluasi model regresi, kami menggunakan metrik utama termasuk R-kuadrat atau koefisien determinasi (R 2 ), kesalahan absolut rata-rata (MAE), dan kesalahan akar-rata-kuadrat (RMSE). Pendekatan komprehensif ini memungkinkan kami untuk menyempurnakan model kami untuk meningkatkan akurasi prediktif dan generalisasi yang lebih baik ke data baru.
2.6 Tes Akhir
Pada tahap ini, model dievaluasi menggunakan set pengujian yang terdiri dari 30% dari total set data. Subset ini ditahan selama fase pelatihan dan validasi untuk memastikan evaluasi yang tidak bias. Kinerja model akhir dinilai berdasarkan data yang tidak terlihat ini, yang memberikan wawasan tentang kemampuannya untuk menggeneralisasi dan secara akurat memprediksi energi aktivasi untuk reaksi yang melibatkan material MXene. Hasil ini penting untuk memverifikasi akurasi dan keandalan prediktif model.
3 Hasil dan Pembahasan
3.1 Distribusi dan Analisis Energi Aktivasi
Sebelum memperkirakan dan menganalisis energi aktivasi, sangat penting untuk memahami distribusi dan variasinya di seluruh kumpulan data, karena ini memberikan wawasan utama tentang potensi MXenes untuk aplikasi dalam katalisis. Gambar 5 menawarkan analisis terperinci tentang distribusi kepadatan dan frekuensi energi aktivasi untuk reaksi disosiasi H 2 O dan OH, serta reaksi asosiasi yang mengarah pada pembentukan H 2 dan CO 2 pada permukaan MXene. Gambar 5a menyajikan distribusi kepadatan energi aktivasi untuk reaksi disosiasi H 2 O dan OH dan untuk reaksi asosiasi yang menghasilkan H 2 dan CO 2 . Energi aktivasi untuk H 2 O (biru) terutama terkonsentrasi pada nilai rendah, memuncak sekitar 0,44 eV, menunjukkan bahwa reaksi disosiasi H 2 O umumnya membutuhkan lebih sedikit energi dibandingkan dengan adsorbat lainnya. Untuk OH (hijau), distribusinya memuncak tajam sekitar 0,74 eV, yang menunjukkan rentang energi aktivasi yang sempit, yang berarti bahwa reaksi disosiasi terkait OH menunjukkan kebutuhan energi yang relatif seragam. Sebaliknya, distribusi energi aktivasi untuk H 2 (merah) jauh lebih luas, mencakup lebih dari 3 eV, yang mencerminkan variabilitas signifikan dalam energi yang dibutuhkan untuk asosiasi H 2 , kemungkinan karena interaksi permukaan yang berbeda atau beberapa jalur reaksi. Demikian pula, untuk CO 2 (sian), energi aktivasi juga tersebar cukup luas, mencapai puncak sekitar 2,3 eV dengan ekor panjang yang melampaui 3 eV, yang menunjukkan bahwa disosiasi CO 2 pada permukaan MXene relatif intensif energi dan menunjukkan variabilitas yang cukup besar. Tumpang tindih antara distribusi ini menunjukkan bahwa beberapa energi aktivasi untuk berbagai adsorbat berada dalam rentang yang sama, yang mungkin menyiratkan persaingan untuk situs adsorpsi atau jalur reaksi bersama.
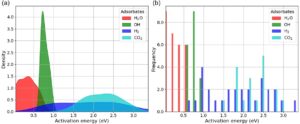
Gambar 5b lebih lanjut mendukung pengamatan ini dengan menampilkan distribusi frekuensi energi aktivasi, yang menawarkan hitungan kejadian yang diskret. Nilai energi aktivasi untuk H 2 O padat di bawah 0,5 eV sedangkan untuk OH terkelompok antara 0,5 dan 1 eV. Dalam kasus reaksi terhadap H 2 , energi aktivasi menunjukkan distribusi yang lebih luas, dengan kejadian yang sering sekitar 0,5–1 dan 2–3 eV, yang menunjukkan beberapa jalur reaksi atau variasi dalam interaksi permukaan MXene. Demikian pula, untuk reaksi oksidasi CO menjadi CO 2 , energi aktivasi mencakup rentang 1,5–3 eV.
Sebelumnya, ditemukan bahwa MXenes dengan konfigurasi d2 umumnya menunjukkan hambatan energi yang lebih rendah terhadap disosiasi H2O , diikuti oleh d3 dan kemudian oleh d4 , dengan MXenes berbasis tungsten menjadi yang paling tidak efektif dari logam yang dianalisis [ 56 ]. Wawasan ini menekankan pentingnya penyempurnaan komposisi dalam merancang MXenes yang disesuaikan untuk reaksi katalitik tertentu. Sementara energi aktivasi ∼0 eV secara teoritis akan mewakili skenario ideal untuk reaksi spontan, kasus seperti itu sangat jarang terjadi dalam sistem nyata [ 77 ]. Faktanya, hambatan aktivasi dalam rentang tertentu lebih disukai untuk aplikasi dalam praktik, karena mereka menyeimbangkan reaktivitas dan selektivitas, memastikan katalisis yang terkontrol dan efisien [ 20 ].
3.2 Menjelajahi Pemilihan dan Analisis Fitur
Pemilihan fitur memainkan peran penting dalam meningkatkan kinerja dan interpretabilitas model dengan mengidentifikasi variabel yang paling berdampak sambil mengurangi kompleksitas dan mengurangi overfitting (Gambar 6 ). Menggunakan Eliminasi Fitur Rekursif dengan Hutan Acak, dua fitur utama diidentifikasi dari kumpulan awal lebih dari 100 sebagai set optimal untuk memprediksi energi aktivasi reaksi yang ada dalam set data (reaksi disosiasi H 2 O dan OH, dan reaksi asosiasi yang mengarah ke H 2 dan CO 2 ). Fitur pertama yang dipilih adalah energi reaksi, yang mewakili perubahan energi yang terjadi selama reaksi, dari reaktan menjadi produk. Fitur ini penting karena berkorelasi langsung dengan kelayakan termodinamika dan stabilitas perantara reaksi, yang memengaruhi persyaratan energi aktivasi. Fitur kedua adalah LogP reaktan, logaritma koefisien partisi oktanol/air, yang menunjukkan bagaimana reagen mendistribusikan dirinya sendiri antara fase hidrofobik (larut dalam lemak) dan hidrofilik (larut dalam air). Meskipun sifat ini mungkin mengejutkan, ia berkontribusi terhadap ukuran sifat hidrofilik molekul penyerap, karenanya secara tidak langsung memengaruhi perilaku penyerapan.
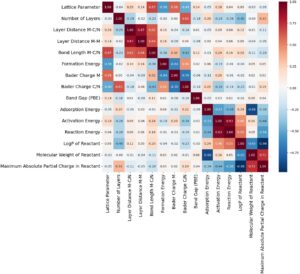
Sifat hidrofilik suatu molekul dapat bergantung pada interaksi antara polaritas, ikatan hidrogen, ukuran molekul, bentuk molekul, dan muatannya. Oleh karena itu, LogP merupakan deskriptor tunggal, yang memiliki kemampuan untuk memperhitungkan berbagai sifat fisikokimia dalam parameter yang sama, yang dipilih menggunakan RFE di antara deskriptor lain yang dapat digunakan untuk mengidentifikasi molekul utama dalam setiap reaksi dalam penelitian ini, seperti berat molekul, luas permukaan polar topologi (TPSA), kesamaan molekul sidik jari yang dihitung menurut apa yang disebut kerapatan Morgan, muatan parsial absolut maksimum, jumlah elektron radikal, dan jumlah elektron valensi. Proses RFE, yang dipandu oleh kesalahan absolut rata-rata sebagai metrik evaluasi, menggabungkan validasi silang untuk memastikan kekokohan pemilihan fitur. Dengan secara sistematis menghilangkan fitur yang tidak relevan atau berlebihan, pendekatan ini meningkatkan akurasi prediktif dan generalisasi model.
Fitur-fitur tambahan disertakan dalam Gambar 6 untuk menyelidiki serangkaian hubungan yang lebih luas dalam kumpulan data, yang akan memberikan pemahaman yang lebih mendalam tentang sistem secara keseluruhan. Fitur-fitur tambahan ini mencakup berat molekul spesies adsorbat utama (H 2 O, OH, H 2 dan CO 2 ) yang memengaruhi interaksinya dengan MXenes; muatan parsial absolut maksimum dari spesies adsorbat utama, yang terkait dengan reaktivitasnya; dan karakteristik khusus MXene, seperti energi pembentukan dan energi adsorpsi reaktan, yang menggambarkan stabilitas dan kekuatan interaksi antara MXenes dan reaktan.
Jarak antara atom dalam struktur MXene juga dipertimbangkan, karena dapat dikaitkan dengan distribusi elektronik permukaan MXene. Menyertakan fitur-fitur tambahan ini, di samping yang utama, memungkinkan eksplorasi yang lebih halus dari interaksi kompleks antara sifat-sifat MXene dan adsorbat. Pendekatan ini memberikan pemahaman yang lebih komprehensif tentang faktor-faktor yang memengaruhi prediksi energi aktivasi, memastikan bahwa model tersebut tidak hanya menangkap prediktor yang paling penting tetapi juga pandangan yang lebih luas tentang struktur kumpulan data dan interaksi rumit antara sifat-sifat bahan dan perilaku katalitiknya. Untuk lebih memahami hubungan antara fitur-fitur, termasuk yang utama yang dipilih, kami menganalisis matriks korelasi, yang divisualisasikan sebagai peta panas pada Gambar 6. Koefisien korelasi berkisar dari + 1 hingga −1, di mana + 1 (ditunjukkan dengan warna merah tua) mewakili korelasi positif sempurna, yang berarti bahwa ketika satu fitur meningkat, yang lain juga meningkat secara proporsional. Sebaliknya, −1 (berwarna biru tua) menunjukkan korelasi negatif sempurna, di mana peningkatan pada satu fitur berkorespondensi dengan penurunan pada fitur lainnya. Nilai mendekati 0 (ditunjukkan dengan warna putih) menunjukkan tidak ada hubungan linear yang signifikan antara variabel. Peta panas ini mengungkap pola penting yang membantu mengidentifikasi saling ketergantungan dan redundansi di antara fitur. Fitur dengan korelasi kuat dapat memberikan informasi yang tumpang tindih, sementara fitur dengan korelasi lemah memberikan kontribusi lebih independen. Analisis ini mendukung relevansi fitur yang dipilih, memastikan fitur tersebut tidak hanya menambah nilai secara independen tetapi juga selaras dengan prinsip fisika dan kimia yang mendasari sistem.
Seperti yang diamati pada Gambar 6 , energi reaksi menunjukkan korelasi positif yang kuat dengan energi aktivasi, yaitu, reaksi yang lebih eksotermik/endotermik cenderung memiliki energi aktivasi yang lebih rendah/lebih tinggi. Pengamatan ini mendukung prinsip Brønsted–Evans–Polanyi dalam katalisis heterogen, yang menunjukkan bahwa energi reaksi dan energi aktivasi terkait erat dalam proses katalitik [ 78 , 79 ]. Energi adsorpsi reaktan memiliki korelasi negatif yang kuat dengan energi aktivasi, yang menunjukkan bahwa adsorpsi reaktan yang lebih kuat ke permukaan katalis cenderung menghasilkan hambatan energi yang lebih tinggi [ 80 ].
Korelasi negatif yang kuat antara LogP dan muatan parsial maksimum dalam reaktan menunjukkan bahwa molekul yang lebih hidrofobik cenderung memiliki muatan parsial yang lebih rendah, kemungkinan karena tidak adanya gugus fungsi yang sangat polar. Namun, mengingat ukuran molekul yang diteliti kecil, tren ini mungkin dibatasi oleh variabilitas yang terbatas dalam strukturnya. Demikian pula, meskipun berat molekul menunjukkan korelasi negatif dengan LogP, hal ini tidak selalu menyiratkan hubungan langsung antara ukuran dan sifat hidrofobik, karena polaritas molekul dipengaruhi oleh gugus fungsi lokal dan distribusi muatan keseluruhan.
Harap perhatikan bahwa menganalisis korelasi, seperti yang diamati antara berat molekul dan energi adsorpsi, dapat menyesatkan jika konteks yang lebih luas diabaikan atau jika hanya sekumpulan spesies terbatas yang dipertimbangkan. Mekanisme adsorpsi sangat kompleks dan, oleh karena itu, analisis tersebut harus dilakukan dengan hati-hati.
3.3 Pemodelan Prediktif Energi Aktivasi
Kami memulai penelitian kami dengan menggunakan regresi linier untuk memprediksi energi aktivasi. Hasil awal dari validasi silang menghasilkan nilai R 2 sebesar 0,79, yang menunjukkan tingkat kemampuan prediktif yang sedang. Meskipun demikian, kinerja model regresi linier dapat dibatasi oleh ketidakmampuannya untuk menangkap hubungan non-linier yang kompleks antara fitur dan energi aktivasi. Oleh karena itu, metode pembelajaran mesin lainnya, seperti regresor hutan acak, SVM, dan jaringan saraf, juga diuji karena metode tersebut lebih mampu menangani kompleksitas data dan sangat efektif dalam menangkap interaksi non-linier yang sering terlewatkan oleh model linier.
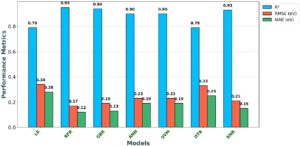
Hasil validasi silang untuk setiap model yang dipelajari dalam karya ini dirangkum dalam Gambar 7. Model non-linier, kecuali DTR, menunjukkan tingkat akurasi prediktif yang lebih tinggi. Lebih jauh, kami mengevaluasi berbagai model menggunakan berbagai status acak untuk pemisahan data antara data yang digunakan untuk validasi silang dan set uji akhir, dan hasilnya tetap konsisten.
Kami mengembangkan model RFR dan mengevaluasi kinerjanya menggunakan validasi silang 5 kali lipat. Metrik kinerja, termasuk R 2 , RMSE, dan MAE, beserta deviasi standarnya, dirangkum dalam Tabel 1 . Deviasi standar yang rendah semakin menegaskan stabilitas kinerja model. Secara keseluruhan, model RFR menunjukkan kemampuan prediktif yang kuat, memodelkan energi aktivasi secara efektif, dan menggeneralisasi dengan baik ke data baru, menjadikannya pilihan yang andal.
| Model | Rata-rata R 2 | Rata-rata RMSE (eV) | Rata-rata MAE (eV) |
|---|---|---|---|
| Bahasa Indonesia: LR | 0,79 ± 0,13 | 0,34 ± 0,02 | 0,28 ± 0,03 |
| RFR | 0,95 ± 0,04 | 0,17 ± 0,06 | 0,12 ± 0,04 |
| Inggris Raya | 0,94 ± 0,04 | 0,19 ± 0,06 | 0,13 ± 0,03 |
| JAM | 0,90 ± 0,06 | 0,23 ± 0,02 | 0,19 ± 0,02 |
| Bahasa Indonesia: SVM | 0,90 ± 0,06 | 0,23 ± 0,02 | 0,19 ± 0,02 |
| DTR | 0,79 ± 0,17 | 0,33 ± 0,16 | 0,25 ± 0,10 |
| KNR | 0,93 ± 0,03 | 0,21 ± 0,06 | 0,15 ± 0,04 |
Bahasa Indonesia: Untuk mengoptimalkan model, kami menggunakan RandomizedSearchCV milik scikit-learn [ 81 ], sebuah teknik yang mengidentifikasi hiperparameter terbaik dengan mengambil sampel secara acak dari nilai yang telah ditetapkan sebelumnya, daripada menguji semua kemungkinan kombinasi. Pendekatan ini membantu untuk mengeksplorasi ruang hiperparameter secara efisien sambil mengurangi biaya komputasi. Deskripsi yang lebih terperinci tentang bagaimana metode ini diterapkan dalam pekerjaan kami, bersama dengan parameter terbaik untuk model, disediakan dalam Informasi Pendukung. Proses pengoptimalan menghasilkan peningkatan kinerja, meningkatkan akurasi dan stabilitas model dalam memprediksi energi aktivasi. Kami mengevaluasi kinerja model menggunakan metrik utama: R 2 ; MAE, yang menghitung besarnya rata-rata kesalahan antara nilai yang diprediksi dan aktual; dan RMSE, yang memberikan bobot lebih besar pada kesalahan yang lebih besar dengan mengevaluasi akar kuadrat dari rata-rata perbedaan kuadrat antara nilai yang diprediksi dan aktual [ 82 ].
Hiperparameter yang dioptimalkan adalah: {‘n_estimators’: 100, ‘min_samples_split’: 2, ‘min_samples_leaf’: 2, ‘max_features’: ‘sqrt’, ‘max_depth’: 30, ‘bootstrap’: False}. Dalam kondisi ini, model mencapai mean cross-validation R 2 sebesar 0,95 ± 0,04, mean RMSE sebesar 0,17 ± 0,06 eV, dan mean MAE sebesar 0,12 ± 0,04 eV. Hasil ini menunjukkan bahwa model tersebut berkinerja baik, menunjukkan R 2 yang tinggi dan RMSE dan MAE yang rendah, yang menyoroti akurasi model RFR dan kemampuannya yang kuat untuk memprediksi energi aktivasi secara efektif. Setelah optimasi dan CV, kami menguji model RFR pada kumpulan data independen, yang menghasilkan hasil yang sangat baik.
Gambar 8 terdiri dari dua subplot untuk pengujian akhir: (a) plot sebar yang membandingkan energi aktivasi yang diprediksi vs. aktual dan (b) histogram residual (Nilai Aktual – Prediksi). Dalam subplot (a), titik-titik data sangat selaras dengan garis diagonal “Perfect Fit”, yang menunjukkan bahwa prediksi model sangat akurat. Kedekatan titik-titik dengan garis regresi lebih lanjut menyoroti korelasi yang kuat antara nilai aktual dan prediksi, didukung oleh koefisien determinasi yang tinggi R 2 = 0,97, RMSE = 0,17 eV dan MAE = 0,14 eV, yang menunjukkan kemampuan prediktif yang sangat baik. Ini menunjukkan bahwa model tersebut secara efektif menangkap deskriptor utama yang memengaruhi energi aktivasi dan dapat digunakan dengan andal untuk prediksi pada permukaan MXene. Subplot (b) menyajikan residual, divisualisasikan sebagai histogram, yang menunjukkan perbedaan antara energi aktivasi aktual dan prediksi. Sebagian besar residual berpusat di sekitar 0 eV, yang mengonfirmasi kinerja prediktif model yang kuat, dengan sebagian besar nilai berada di antara −0,3 dan 0,3 eV. Distribusi yang ketat ini menunjukkan bahwa kesalahan prediksi minimal dan terdistribusi secara merata. Besaran residual yang rendah ini memperkuat keandalan model, menjadikannya alat yang tangguh untuk prediksi energi aktivasi dalam aplikasi katalisis.
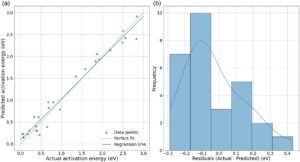
Sementara residual pada Gambar 8b sebagian besar terkonsentrasi dalam ±0,3 eV, dengan hanya satu residual yang berada di luar rentang ini, yaitu permukaan Ta 2 C untuk reaksi CO + O → CO 2 , dengan residual sebesar 0,43. Ini adalah persentase kecil dari set pengujian (∼3,5%), dengan semua contoh lainnya mengikuti distribusi kesalahan normal. Dispersi ini menunjukkan bahwa penyimpangan mungkin timbul dari lingkungan lokal yang kompleks atau kurang terwakili dalam komposisi MXene tertentu. Meskipun contoh seperti itu ada, jumlah dan distribusi yang sangat terbatas tidak menunjukkan bias sistematis dalam model. Yang penting, kehadiran mereka memperkuat nilai perluasan set data dan menggabungkan deskriptor yang lebih bernuansa dalam pekerjaan mendatang, khususnya untuk menangkap variasi elektronik atau struktural yang lebih halus yang memengaruhi prediksi energi aktivasi.
Kami juga mengeksplorasi penggunaan model GBR, yang menunjukkan kinerja yang baik dalam memprediksi energi aktivasi, menurut metrik kinerja validasi silang berikut: R 2 = 0,94 ± 0,04, RMSE = 0,19 ± 0,06 eV, dan MAE = 0,13 ± 0,03 eV. Namun, ketika membandingkan kinerja model GBR dengan model RFR, kami menemukan bahwa model RFR sedikit mengungguli GBR dalam hal R 2 . Akibatnya, model RFR dipilih untuk analisis lebih lanjut, khususnya untuk mengeksplorasi pentingnya fitur dalam sisa studi ini.
Selain itu, kami mengevaluasi algoritme lain, termasuk ANN, SVM, DTR, dan KNR, untuk membandingkan kinerjanya. Berdasarkan metrik CV yang dirangkum dalam Tabel 1 , RFR secara konsisten mengungguli semua model lainnya. Sebaliknya, selain LR, DTR menunjukkan mean R 2 terendah , 0,79 ± 0,17, dan variabilitas tertinggi dalam kinerja. Ia juga mencatat RMSE tertinggi sebesar 0,33 ± 0,16 eV dan MAE sebesar 0,25 ± 0,10 eV, menjadikannya model yang paling tidak dapat diandalkan dan akurat. Secara keseluruhan, RFR muncul sebagai model yang paling dapat diandalkan dan akurat untuk memprediksi energi aktivasi. Ia secara efektif menangkap interaksi nonlinier yang kompleks dan digeneralisasi dengan baik di seluruh kumpulan data, sebagaimana dibuktikan oleh validasi silang 5 kali lipat dan set pengujian. Sebuah studi baru-baru ini, yang menggunakan pembelajaran mesin untuk memprediksi energi adsorpsi berbagai molekul (H 2 O, CO 2 , H 2 , CO, O 2 , OH, O, dan H) pada MXenes, juga mengidentifikasi RFR sebagai algoritma yang paling efektif, yang selanjutnya mengonfirmasi potensinya dalam domain ini [ 50 ]. Khususnya, RFR mengungguli pendekatan alternatif seperti gradient boosting, jaringan saraf, dan SVM. Arsitektur ensemble dan ketahanan inheren terhadap overfitting membuatnya sangat cocok untuk dataset heterogen seperti yang digunakan dalam studi ini. Sementara RFR menawarkan kinerja prediktif yang kuat, interpretabilitasnya yang terbatas pada tingkat prediksi individu dapat menghalangi ekstraksi wawasan mekanistik yang terperinci. Untuk mengatasi hal ini, analisis SHapley Additive exPlanations (SHAP) [ 83 , 84 ] dilakukan untuk memahami bagaimana fitur spesifik memengaruhi prediksi individu (bagian berikutnya).
Studi ini berfokus pada penggunaan ML untuk memprediksi energi aktivasi untuk berbagai reaksi yang melibatkan air, hidroksil, hidrogen, dan karbon dioksida pada permukaan MXene. MXene, yang dikenal karena sifat katalitiknya yang serbaguna, dirancang khusus untuk berbagai reaksi kimia, khususnya yang penting dalam teknologi energi berkelanjutan. Studi ini mengeksplorasi hubungan antara sifat katalitik permukaan, seperti energi adsorpsi dan kemampuan transfer muatan, dan energi aktivasi. Meskipun kumpulan data terbatas, model pembelajaran mesin menunjukkan akurasi prediktif yang menjanjikan, dengan rencana untuk memperluas kumpulan data dan menggabungkan model tambahan untuk kinerja yang lebih baik. Pekerjaan di masa mendatang akan difokuskan pada peningkatan kumpulan data melalui simulasi komputasional, termasuk lebih banyak reaksi dan spesies permukaan, dan meningkatkan interpretabilitas dan keandalan model melalui metode seperti simulasi DFT. Upaya ini bertujuan untuk meningkatkan kerangka kerja prediktif dan mendukung pengembangan solusi katalitik umum yang dapat diskalakan.
Dalam studi Hutton et al . [ 85 ] yang berfokus pada prediksi energi aktivasi untuk reaksi kimia pada permukaan logam, penulis menggunakan RFR, SVR, dan GBR untuk memprediksi energi aktivasi untuk reaksi yang melibatkan molekul yang mengandung C, O, dan H pada permukaan logam transisi. Mereka melaporkan metrik kinerja berikut: RFR dengan kesalahan absolut rata-rata 0,17 eV, GBR dengan MAE 0,15 eV, dan SVR dengan MAE 0,17 eV. Sebagai perbandingan, pekerjaan kami mengambil pendekatan serupa dengan menggunakan model pembelajaran mesin untuk memprediksi energi aktivasi, tetapi dengan fokus pada MXenes. Namun, hasil kami melampaui metrik kinerja yang disajikan oleh Hutton et al., dengan MAE 0,12 eV. Ini mendukung bahwa model kami sangat cocok untuk memprediksi energi aktivasi pada permukaan MXene, mungkin karena sifat unik MXenes dan pendekatan khusus yang kami adopsi untuk memperhitungkan karakteristik permukaannya. Kedua studi tersebut menyoroti keunggulan model non-linier, seperti RFR, dibandingkan regresi linier tradisional, yang kesulitan menangkap kompleksitas interaksi dalam sistem katalitik. Persamaan antara karya kami dan karya Hutton dkk., menekankan peran ML yang semakin besar dalam mempercepat pemahaman sistem katalitik yang kompleks.
Temuan dari pekerjaan kami menunjukkan bahwa model ML, khususnya metode ensemble seperti RFR dan GBR, dapat secara efektif memprediksi energi aktivasi bahkan ketika diterapkan pada kumpulan data yang relatif kecil (92 titik data). Sementara kumpulan data yang lebih kecil sering dianggap sebagai keterbatasan dalam pembelajaran mesin, kami menyadari bahwa dengan pemilihan model dan penanganan data yang cermat, prediksi yang efektif masih dapat dicapai. Untuk mengurangi overfitting dan memastikan ketahanan model kami, kami menggunakan beberapa strategi, termasuk validasi silang 5 kali lipat, teknik regularisasi, dan pengoptimalan hiperparameter melalui RandomizedSearchCV. Studi yang dilaporkan tahun lalu mendukung validitas pendekatan pembelajaran mesin, bahkan ketika diterapkan pada kumpulan data kecil dalam penelitian katalisis. Misalnya, Taniike et al. [ 86 ] mengembangkan metode rekayasa fitur otomatis yang mengekstrak deskriptor relevan tanpa pengetahuan khusus domain sebelumnya dan berhasil menerapkannya pada tiga sistem katalitik yang berbeda dengan data terbatas. Hasil mereka menunjukkan bahwa model ML yang akurat masih dapat dikembangkan di bawah kendala tersebut. [ 87 ] meninjau penerapan ML dalam katalisis dan menekankan bahwa bahkan kumpulan data kecil dan spesifik domain dapat menghasilkan wawasan berharga jika digabungkan dengan pemilihan algoritma yang tepat dan prinsip penanganan data yang konsisten. Demikian pula, Chen et al. [ 88 ] memperkenalkan alur kerja berorientasi data kecil untuk aktivasi propana katalitik dan menemukan bahwa model ensemble seperti Random Forest dan CatBoost sangat efektif dalam skenario data kecil. Ketiga contoh ini memperkuat bahwa dengan pemilihan model dan kurasi data yang cermat, seperti yang diterapkan dalam studi kami, keandalan dan interpretabilitas dapat dipertahankan secara efektif.
3.4 Analisis Pentingnya Fitur dan SHAP
Analisis kepentingan fitur adalah teknik penting yang digunakan untuk memahami bagaimana fitur atau variabel individual memengaruhi hasil prediksi dalam sebuah model. Ini membantu menentukan faktor mana yang memiliki dampak terbesar pada keakuratan model, memungkinkan pemahaman yang lebih baik tentang data yang mendasarinya dan hubungannya [ 89 , 90 ]. Dalam studi kami, kami menggunakan kepentingan permutasi fitur, sebuah metode yang menilai kontribusi setiap fitur dengan mengacak nilainya secara acak dan mengukur perubahan yang dihasilkan dalam kinerja model. Penurunan kinerja yang nyata menunjukkan bahwa fitur tersebut sangat penting, sementara perubahan minimal menunjukkan fitur tersebut memiliki relevansi yang lebih rendah terhadap prediksi model. Teknik ini dapat diterapkan pada semua jenis model dan menawarkan wawasan tentang faktor-faktor paling berpengaruh yang mendorong hasil model. Gambar 9 menunjukkan proses ini menggunakan RFR, di mana penurunan R 2 membantu mengukur nilai prediktif setiap fitur.
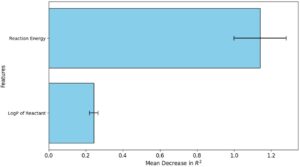
Energi reaksi muncul sebagai fitur paling signifikan dalam model, menunjukkan penurunan rata-rata tertinggi dalam R 2 , yang menunjukkan pengaruh dominannya pada akurasi prediktif model. Korelasi yang kuat antara kedua variabel ini menyoroti peran penting yang dimainkan oleh sifat termodinamika dalam menentukan hambatan energi suatu reaksi, sementara energi aktivasi secara khusus mengatur kinetika dengan mendikte hambatan energi yang harus diatasi agar reaksi dapat berlanjut. Jadi, seperti yang diharapkan, menghilangkan energi reaksi dari model akan menyebabkan pengurangan yang nyata dalam kemampuan prediktifnya, yang menggarisbawahi pentingnya energi reaksi dalam pemodelan energi aktivasi yang akurat. Energi reaksi merangkum perubahan termodinamika yang terjadi selama transformasi kimia reaktan menjadi produk. Ini berfungsi sebagai indikator fundamental dari efisiensi katalitik suatu sistem, yang mencerminkan energi yang dilepaskan atau dikonsumsi dalam reaksi. Pentingnya energi reaksi dalam konteks ini menunjukkan interaksi substansial antara termodinamika dan energi aktivasi, yang konsisten dengan prinsip Brønsted–Evans–Polanyi [ 91 , 92 – 93 ]. Prinsip ini menyatakan bahwa energi aktivasi suatu reaksi biasanya terkait dengan energi reaksinya, yaitu, reaksi yang lebih eksergonik (menguntungkan secara termodinamika) umumnya menunjukkan hambatan aktivasi yang lebih rendah. Hubungan ini menekankan bahwa keuntungan termodinamika suatu reaksi secara langsung memengaruhi parameter kinetiknya, menawarkan pemahaman yang lebih dalam tentang perilaku katalitik yang mendasarinya.
Selain pentingnya permutasi fitur, analisis SHAP juga dilakukan untuk menganalisis pentingnya fitur dan dampaknya pada output. Menurut hasil yang disajikan dalam Gambar 10 , energi reaksi adalah fitur yang paling penting, yang memvalidasi hasil pentingnya permutasi fitur sebelumnya. Selain itu, menurut nilai SHAP rata-rata, perubahan energi reaksi menggerakkan penghalang aktivasi yang diprediksi sekitar dua kali lebih banyak, rata-rata, daripada perubahan LogP. Menurut nilai SHAP yang disajikan dalam grafik di bawah ini pada Gambar 10 , untuk energi reaksi (baris atas), energi reaksi yang lebih rendah (biru) mengelompok di sebelah kiri (SHAP negatif), yang berarti bahwa mereka cenderung mendorong penghalang yang diprediksi model ke bawah. Sedangkan, energi reaksi yang lebih tinggi (merah) mengelompok di sebelah kanan (SHAP positif), yang berarti bahwa mereka cenderung menghasilkan energi aktivasi yang lebih tinggi. Untuk LogP reaktan (baris bawah), nilai LogP yang lebih rendah (biru) cenderung memberikan SHAP negatif, yang berarti bahwa, dalam model, mereka sedikit menurunkan penghalang yang diprediksi. Sedangkan LogP yang tinggi (merah) cenderung memberikan SHAP positif (meningkatkan energi aktivasi yang diprediksi).
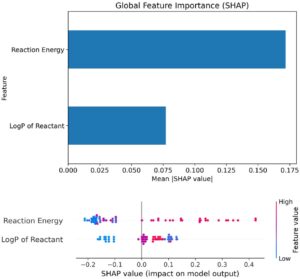
Ini sangat berguna untuk perhitungan prinsip-prinsip pertama energi aktivasi, karena energi reaksi secara signifikan lebih murah untuk dihitung secara komputasional. Mengetahui bahwa hubungan seperti BEP berlaku, energi reaksi kemudian dapat digunakan untuk menilai (setidaknya sebagai pendahuluan) potensi katalitik permukaan MXene. Sementara ada pengecualian dalam kasus-kasus tertentu [ 94 ], tren umum ini berlaku, memperkuat hubungan antara sifat termodinamika dan kinetika reaksi. Identifikasi energi reaksi sebagai faktor kunci dalam prediksi energi aktivasi selanjutnya menetapkannya sebagai jembatan penting antara aspek termodinamika dan kinetik katalisis, memberikan wawasan berharga ke dalam kinerja katalitik MXene yang dipelajari.
Fajín et al. [ 95 ] mengidentifikasi energi ko-adsorpsi OH dan H, produk disosiasi air, sebagai deskriptor paling signifikan untuk berbagai permukaan, termasuk permukaan monometalik, bimetalik, dan trimetalik, nanotube metalik, dan nanopartikel platinum. Namun, deskriptor ini ditemukan berkorelasi kuat dengan energi reaksi disosiasi air menjadi OH dan H (Persamaan ( 1 )), yang dikecualikan dalam penelitian mereka untuk mencegah multikolinearitas. Jika energi reaksi digunakan sebagai pengganti energi ko-adsorpsi OH dan H, kemungkinan besar akan muncul sebagai deskriptor paling signifikan, seperti yang terlihat dalam pekerjaan kami saat ini. Meskipun lebih menuntut komputasi, energi reaksi menawarkan deskripsi yang lebih komprehensif, yang memperhitungkan keadaan reaksi awal dan akhir.
Nilai LogP dari spesies adsorbat utama (H 2 O, OH, H 2 , dan CO 2 ), ukuran hidrofobisitas senyawa, muncul, secara tak terduga, sebagai fitur terpenting kedua dalam model untuk memprediksi energi aktivasi, meskipun pengaruhnya bersifat sekunder terhadap energi reaksi. Pengamatan ini menunjukkan bahwa sementara hidrofobisitas reaktan, seperti yang ditunjukkan oleh LogP-nya, memang memengaruhi energi aktivasi, ia memberikan efek yang relatif lebih kecil daripada faktor termodinamika seperti energi reaksi. Molekul hidrofobik biasanya menunjukkan perilaku yang berbeda dalam reaksi katalitik, terutama dalam interaksinya dengan permukaan katalitik [ 96 ]. Korelasi sedang antara LogP dan energi aktivasi menyiratkan bahwa hidrofobisitas dapat memengaruhi bagaimana reaktan berinteraksi dengan katalis, sehingga memengaruhi penghalang energi reaksi. Secara khusus, reaktan yang lebih hidrofobik dapat melekat secara berbeda pada permukaan katalis dibandingkan dengan reaktan yang lebih hidrofilik. Perbedaan dalam adsorpsi ini dapat mengubah energi aktivasi, baik dengan menstabilkan atau mendestabilisasi keadaan transisi, tergantung pada sifat interaksi. Efek ini dapat bervariasi tergantung pada permukaan katalitik spesifik dan lingkungan tempat reaksi berlangsung [ 97 ].
Studi kami terutama bertujuan untuk memprediksi energi aktivasi untuk reaksi yang melibatkan MXenes dan spesies H 2 O, OH, CO 2 , dan H 2 (Persamaan ( 1 )–( 4 )) karena pentingnya mereka dalam rute redoks reaksi WGS. Kami fokus pada faktor-faktor yang berhubungan dengan termodinamika dan hidrofobisitas. Pekerjaan masa depan yang menggabungkan fitur struktur elektronik dan efek pelarut dapat lebih meningkatkan akurasi prediktif model kami. Efek pelarut yang dimodelkan menggunakan formalisme pelarut implisit ditemukan secara nyata mempengaruhi energi adsorpsi pada permukaan logam dan dapat relevan, misalnya, dalam reaksi elektrokatalitik [ 98 ]. Hubungan yang diamati antara pentingnya fitur dan kinetika reaksi menunjukkan bahwa pendekatan multi-variabel, yang mengintegrasikan deskriptor termodinamika, kinetik, dan elektronik, dapat memberikan pemahaman yang lebih komprehensif tentang perilaku katalitik. Memperluas pemilihan fitur untuk menyertakan aspek-aspek ini dapat menghasilkan wawasan yang lebih dalam tentang fondasi mekanistik dari variasi energi aktivasi, yang pada akhirnya membantu dalam desain rasional katalis berkinerja tinggi. Selain faktor-faktor ini, distribusi muatan pada permukaan MXene dan jarak antara situs aktif dapat memengaruhi prediksi energi aktivasi dengan memengaruhi adsorpsi reaktan dan stabilisasi keadaan transisi [ 99 ].
Energi adsorpsi, deskriptor utama dalam katalisis heterogen, dapat lebih memperkuat model prediktif dengan mengukur kekuatan interaksi reaktan-katalis. Selain itu, menggabungkan sifat-sifat khusus reaktan, seperti momen dipol, polarisabilitas, dan afinitas elektron, dapat menawarkan wawasan yang lebih mendalam tentang bagaimana karakteristik molekuler membentuk kinerja katalitik. Dalam studi ini, kami menggunakan model prediktif tunggal untuk semua reaksi. Namun, untuk pekerjaan di masa mendatang, mungkin berguna untuk menerapkan pendekatan pembelajaran multitugas atau pemodelan hierarkis. Metode-metode ini dapat membantu memisahkan fitur-fitur yang khusus untuk setiap reaksi dengan lebih baik dari fitur-fitur yang dimiliki bersama di semua reaksi. Ini akan memungkinkan model untuk menangkap tren umum dan pola-pola unik dengan lebih baik, terutama ketika bekerja dengan serangkaian reaksi beragam yang lebih besar atau sistem katalitik yang lebih kompleks.
4 Kesimpulan dan Perspektif
Studi ini menggarisbawahi potensi metode pembelajaran mesin, khususnya teknik ensemble seperti GBR dan RFR, dalam memprediksi energi aktivasi reaksi yang melibatkan MXenes. Di antara model-model ini, RFR menunjukkan kinerja terbaik, mencapai mean cross-validation R 2 yang mengesankan sebesar 0,95, dengan root mean square error sebesar 0,17 eV dan mean absolute error sebesar 0,12 eV. Hasil-hasil ini mengonfirmasi kemanjuran RFR dalam memprediksi energi aktivasi secara akurat, yang menyoroti pentingnya RFR sebagai alat prediksi. Pemilihan fitur mengungkapkan bahwa dua atribut utama, yaitu energi reaksi dan logaritma koefisien partisi reaktan, adalah yang paling berpengaruh dalam menentukan energi aktivasi. Khususnya, energi reaksi diidentifikasi sebagai fitur yang paling penting dalam memengaruhi prediksi model. Wawasan ini sangat penting untuk memahami faktor-faktor utama yang mendorong energi aktivasi reaksi yang terjadi pada permukaan MXene.
Penelitian di masa depan akan mendapat manfaat dari penggabungan titik data tambahan dari perhitungan DFT, memperluas kumpulan data untuk mencakup rentang reaksi elementer yang lebih luas yang relevan dengan proses katalitik. Ini akan memperdalam pemahaman tentang katalisis MXene dan meningkatkan generalisasi dan ketahanan model pembelajaran mesin, yang mengarah ke prediksi yang lebih akurat di berbagai reaksi kimia. Mengintegrasikan fitur tambahan, seperti jarak reaktan ke permukaan, dapat lebih menyempurnakan model ini dengan menangkap efek katalitik yang halus. Dalam konteks ini, perhatian khusus harus diberikan pada molekul pembawa hidrogen seperti metanol (CH 3 OH), formaldehida (H 2 CO), asam format (HCOOH), dan amonia (NH 3 ), yang penting dalam aplikasi terkait energi dan industri. Pilihan reaktan yang beragam akan meningkatkan evaluasi MXenes sebagai katalis, memberikan pemahaman yang lebih komprehensif tentang kinerja katalitiknya.